Understanding Fabry Disease
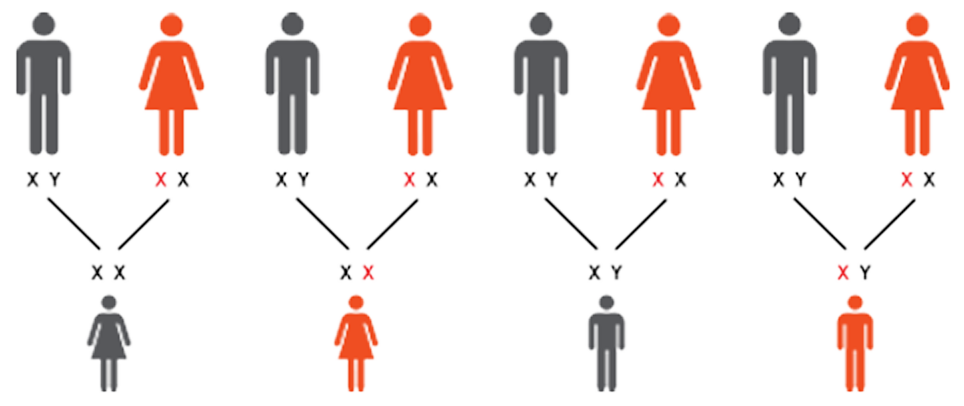
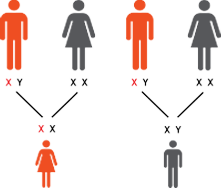
Fabry disease is a progressive, multisystemic, X-linked lysosomal disorder
affecting both males and females.1,2
Fabry disease is caused by mutations in the galactosidase alpha gene (GLA), which encodes the alpha-galactosidase A (alpha-Gal A) enzyme.1,2 These mutations can cause the absence or deficiency of functional alpha-Gal A, which breaks down globotriaosylceramide (GL-3), plasma globotriaosylsphingosine (lyso-Gb3), and other disease substrates in healthy individuals.3,4 When alpha-Gal A is absent or deficient, GL-3, lyso-Gb3, and other disease substrates accumulate, leading to cell damage within affected parts of the individual’s body and causing the various pathologies seen in Fabry disease.3,4 This substrate build-up of GL-3, lyso-Gb3, and other disease substrates can lead to irreversible organ damage.3,4
In addition to the symptoms caused by GL-3 and lyso-Gb3 accumulation, activation of other pathways triggering inflammation may also contribute to the disease process and may be a driver of fibrosis and end organ damage.3,5-7
As a progressive, multisystemic disease, Fabry disease can1:
- Have a devastating impact on people’s lives
- Have a wide spectrum of symptoms
- Present differently in each affected individual
- Be a significant burden regardless of presentation